Lettura Magistrale del Prof. Dr. J. Brettschneider
giugno 28, 2016
Monaco 2016 – ICFTD 2016
novembre 16, 2016Premio AIMFT 2015

Nel corso di “SINdem 2015 Genova” AIMFT ha consegnato a Chiara Cupidi il Premio biennale dell’Associazione.
Il Premio è rivolto al miglior articolo/ricerca scientifica di un giovane ricercatore.
Qui potete trovarne un estratto:
Novel N-terminal domain mutation in prion protein detected in 2 patients diagnosed with frontotemporal lobar degeneration syndrome
Chiara Cupidi, Livia Bernardi, Francesca Frangipane, Maria Anfossi, Maura Gallo, Maria Elena Conidi, Franca Vasso, Rosanna Colao, Gianfranco Puccio, Sabrina Curcio, Maria Mirabelli, Alessandra Clodomiro, Raffaele Di Lorenzo, Nicoletta Smirne, Raffaele Maletta, Amalia C. Bruni
Centro Regionale di Neurogenetica, ASP CZ, Lamezia Terme, Catanzaro, Italy
Neurobiology of Aging. 2014 Nov; 35(11):2657.e7-11.
Riassunto
Le mutazioni del gene PRNP, che codifica per la proteina prionica (PrP), sono solitamente responsabili delle malattie prioniche ereditarie, quali la malattia di Creutzfeldt-Jakob familiare o la sindrome di Gerstmann-Straussler-Schenker. Tuttavia, alcune mutazioni del gene PRNP sono state associate a quadri clinici che possono mimare patologie neurodegenerative differenti dalle consuete malattie prioniche.
In questo studio, abbiamo identificato una nuova mutazione missense del gene PRNP in 2 pazienti affetti da demenza frontotemporale. La nuova mutazione PRNP P39L è sita in corrispondenza del dominio N-terminale della proteina prionica.
Il primo paziente era un uomo di 67 anni affetto da demenza frontotemporale, esordita con un quadro clinico di afasia progressiva non fluente. Il secondo paziente era un uomo di 78 anni
affetto da demenza frontotemporale e parkinsonismo. Nessuno dei due pazienti presentava caratteristiche cliniche che suggerissero una malattia prionica ereditaria. I quadri neuroradiologici sono illustrati nella Figura 1.
I due pazienti sono risultati negativi per mutazioni in altri geni responsabili di demenza familiare (geni MAPT, GRN, C9ORF72, PS1, PS2).
Lo screening genetico di controllo ha dimostrato l’assenza della mutazione PRNP P39L in 200 soggetti di controllo sani e di pari età, suggerendo la potenziale patogenicità di questa variante genetica. L’analisi bioinformatica ha inoltre predetto la variante genetica come “dannosa” e “causa di malattia”.
Le particolari caratteristiche cliniche dei pazienti, non classificabili come malattie da prioni ma come demenza frontotemporale, potrebbero dipendere dalla posizione della mutazione nel dominio N-terminale della proteina prionica, fuori del nucleo amiloide della proteina prionica patologica, anche se ulteriori studi funzionali saranno necessari per determinare se e come questa mutazione
eserciti il suo effetto patogeno. Tuttavia, lo screening genetico del gene della proteina prionica diventa rilevante nelle demenze familiari, in particolare nelle aree geografiche con elevata prevalenza di malattie prioniche ereditarie.
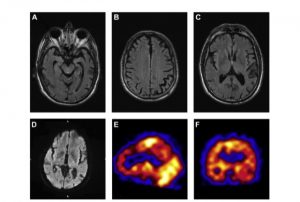
Fig. 1. La risonanza magnetica dell’encefalo ha mostrato in entrambi i pazienti una atrofia cerebrale a carico delle aree temporale, frontale mesiale e parietale posteriore nell’emisfero sinistro (A, B, C), in assenza di lesioni iperintense a carico dei gangli della base. (D). L’esame SPECT cerebrale ha rilevato l’ipoperfusione delle aree corticali parietali e frontali nonchè del lobo temporale sinistra, con un relative risparmio del lobo occipital, del tronco encefalico e del cervelletto (E, F).